

Would A Mismatch Repair Result In More Mutations If Using Newly Sunthesized Strand As A Template
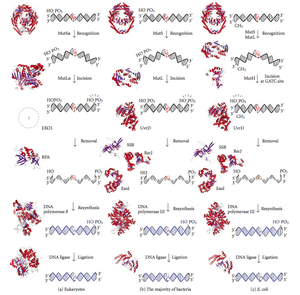
Diagram of DNA mismatch repair pathways. The first cavalcade depicts mismatch repair in eukaryotes, while the 2nd depicts repair in well-nigh leaner. The tertiary column shows mismatch repair, to be specific in Due east. coli.
DNA mismatch repair (MMR) is a system for recognizing and repairing erroneous insertion, deletion, and mis-incorporation of bases that tin arise during DNA replication and recombination, as well as repairing some forms of DNA damage.[one] [2]
Mismatch repair is strand-specific. During DNA synthesis the newly synthesised (girl) strand will commonly include errors. In lodge to begin repair, the mismatch repair machinery distinguishes the newly synthesised strand from the template (parental). In gram-negative bacteria, transient hemimethylation distinguishes the strands (the parental is methylated and daughter is not). However, in other prokaryotes and eukaryotes, the exact mechanism is non clear. It is suspected that, in eukaryotes, newly synthesized lagging-strand Dna transiently contains nicks (before being sealed past DNA ligase) and provides a betoken that directs mismatch proofreading systems to the advisable strand. This implies that these nicks must be present in the leading strand, and evidence for this has recently been found.[3] Recent work[4] has shown that nicks are sites for RFC-dependent loading of the replication sliding clamp, proliferating prison cell nuclear antigen (PCNA), in an orientation-specific mode, such that one face of the donut-shape protein is juxtaposed toward the 3'-OH end at the nick. Loaded PCNA then directs the action of the MutLalpha endonuclease [5] to the daughter strand in the presence of a mismatch and MutSalpha or MutSbeta.
Any mutational event that disrupts the superhelical structure of Deoxyribonucleic acid carries with it the potential to compromise the genetic stability of a cell. The fact that the impairment detection and repair systems are as complex as the replication mechanism itself highlights the importance evolution has fastened to DNA allegiance.
Examples of mismatched bases include a G/T or A/C pairing (see DNA repair). Mismatches are usually due to tautomerization of bases during DNA replication. The harm is repaired by recognition of the deformity caused past the mismatch, determining the template and not-template strand, and excising the wrongly incorporated base and replacing it with the correct nucleotide. The removal process involves more than merely the mismatched nucleotide itself. A few or up to thousands of base pairs of the newly synthesized DNA strand can be removed.
Mismatch repair proteins [edit]
| DNA mismatch repair protein, C-last domain | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 hpms2-atpgs | |||||||||
| Identifiers | |||||||||
| Symbol | DNA_mis_repair | ||||||||
| Pfam | PF01119 | ||||||||
| Pfam clan | CL0329 | ||||||||
| InterPro | IPR013507 | ||||||||
| PROSITE | PDOC00057 | ||||||||
| SCOP2 | 1bkn / SCOPe / SUPFAM | ||||||||
| |||||||||
Mismatch repair is a highly conserved procedure from prokaryotes to eukaryotes. The first testify for mismatch repair was obtained from S. pneumoniae (the hexA and hexB genes). Subsequent work on Eastward. coli has identified a number of genes that, when mutationally inactivated, cause hypermutable strains. The gene products are, therefore, chosen the "Mut" proteins, and are the major active components of the mismatch repair organization. Three of these proteins are essential in detecting the mismatch and directing repair machinery to it: MutS, MutH and MutL (MutS is a homologue of HexA and MutL of HexB).
MutS forms a dimer (MutS2) that recognises the mismatched base of operations on the girl strand and binds the mutated DNA. MutH binds at hemimethylated sites along the daughter DNA, but its action is latent, beingness activated but upon contact by a MutL dimer (MutLtwo), which binds the MutS-DNA complex and acts as a mediator between MutSii and MutH, activating the latter. The Deoxyribonucleic acid is looped out to search for the nearest d(GATC) methylation site to the mismatch, which could exist upwardly to one kb away. Upon activation by the MutS-Deoxyribonucleic acid complex, MutH nicks the girl strand near the hemimethylated site. MutL recruits UvrD helicase (Deoxyribonucleic acid Helicase II) to divide the two strands with a specific iii' to v' polarity. The unabridged MutSHL complex then slides along the DNA in the management of the mismatch, liberating the strand to be excised equally it goes. An exonuclease trails the circuitous and digests the ss-Dna tail. The exonuclease recruited is dependent on which side of the mismatch MutH incises the strand – v' or 3'. If the nick fabricated by MutH is on the 5' terminate of the mismatch, either RecJ or ExoVII (both 5' to 3' exonucleases) is used. If, however, the nick is on the three' finish of the mismatch, ExoI (a 3' to v' enzyme) is used.
The entire procedure ends past the mismatch site - i.e., both the site itself and its surrounding nucleotides are fully excised. The single-strand gap created past the exonuclease can then be repaired by DNA Polymerase Three (assisted by single-strand-bounden protein), which uses the other strand as a template, and finally sealed by Deoxyribonucleic acid ligase. Deoxyribonucleic acid methylase and then rapidly methylates the daughter strand.
MutS homologs [edit]
When spring, the MutStwo dimer bends the DNA helix and shields approximately xx base pairs. It has weak ATPase activity, and binding of ATP leads to the formation of 3rd structures on the surface of the molecule. The crystal structure of MutS reveals that information technology is exceptionally disproportionate, and, while its active conformation is a dimer, but i of the ii halves interacts with the mismatch site.
In eukaryotes, MutS homologs form two major heterodimers: Msh2/Msh6 (MutSα) and Msh2/Msh3 (MutSβ). The MutSα pathway is involved primarily in base substitution and pocket-sized-loop mismatch repair. The MutSβ pathway is as well involved in small-loop repair, in addition to large-loop (~x nucleotide loops) repair. However, MutSβ does non repair base of operations substitutions.
MutL homologs [edit]
MutL also has weak ATPase action (it uses ATP for purposes of movement). It forms a complex with MutS and MutH, increasing the MutS footprint on the DNA.
However, the processivity (the distance the enzyme tin move forth the Dna before dissociating) of UvrD is simply ~40–50 bp. Because the altitude between the nick created by MutH and the mismatch tin average ~600 bp, if there is non another UvrD loaded the unwound section is so free to re-anneal to its complementary strand, forcing the process to beginning over. Still, when assisted by MutL, the rate of UvrD loading is greatly increased. While the processivity (and ATP utilisation) of the individual UvrD molecules remains the same, the total effect on the DNA is additional considerably; the DNA has no chance to re-anneal, as each UvrD unwinds 40-50 bp of DNA, dissociates, and so is immediately replaced by another UvrD, repeating the process. This exposes large sections of DNA to exonuclease digestion, allowing for quick excision (and later replacement) of the incorrect DNA.
Eukaryotes have five MutL homologs designated as MLH1, MLH2, MLH3, PMS1, and PMS2. They form heterodimers that mimic MutL in E. coli. The human homologs of prokaryotic MutL grade three complexes referred to as MutLα, MutLβ, and MutLγ. The MutLα complex is made of MLH1 and PMS2 subunits, the MutLβ heterodimer is made of MLH1 and PMS1, whereas MutLγ is fabricated of MLH1 and MLH3. MutLα acts as an endonuclease that introduces strand breaks in the daughter strand upon activation past mismatch and other required proteins, MutSα and PCNA. These strand interruptions serve as entry points for an exonuclease activity that removes mismatched Deoxyribonucleic acid. Roles played by MutLβ and MutLγ in mismatch repair are less-understood.
MutH: an endonuclease present in Eastward. coli and Salmonella [edit]
MutH is a very weak endonuclease that is activated one time bound to MutL (which itself is bound to MutS). It nicks unmethylated Dna and the unmethylated strand of hemimethylated DNA but does not nick fully methylated Dna. Experiments have shown that mismatch repair is random if neither strand is methylated.[ citation needed ] These behaviours led to the proposal that MutH determines which strand contains the mismatch. MutH has no eukaryotic homolog. Its endonuclease function is taken upwardly past MutL homologs, which have some specialized five'-3' exonuclease activity. The strand bias for removing mismatches from the newly synthesized daughter strand in eukaryotes may be provided by the free 3' ends of Okazaki fragments in the new strand created during replication.
PCNA β-sliding clamp [edit]
PCNA and the β-sliding clamp acquaintance with MutSα/β and MutS, respectively. Although initial reports suggested that the PCNA-MutSα complex may enhance mismatch recognition,[half-dozen] it has been recently demonstrated[7] that in that location is no credible change in affinity of MutSα for a mismatch in the presence or absence of PCNA. Furthermore, mutants of MutSα that are unable to interact with PCNA in vitro showroom the capacity to bear out mismatch recognition and mismatch excision to near wild blazon levels. Such mutants are lacking in the repair reaction directed by a v' strand suspension, suggesting for the starting time time MutSα function in a post-excision step of the reaction.
Clinical significance [edit]
Inherited defects in mismatch repair [edit]
Mutations in the homo homologues of the Mut proteins affect genomic stability, which can event in microsatellite instability (MSI), implicated in some human being cancers. In specific, the hereditary nonpolyposis colorectal cancers (HNPCC or Lynch syndrome) are attributed to dissentious germline variants in the genes encoding the MutS and MutL homologues MSH2 and MLH1 respectively, which are thus classified as tumour suppressor genes. One subtype of HNPCC, the Muir-Torre Syndrome (MTS), is associated with pare tumors. If both inherited copies (alleles) of a MMR gene bear damaging genetic variants, this results in a very rare and severe condition: the mismatch repair cancer syndrome (or constitutional mismatch repair deficiency, CMMR-D), manifesting as multiple occurrences of tumors at an early historic period, often colon and brain tumors.[eight]
Epigenetic silencing of mismatch repair genes [edit]
Sporadic cancers with a DNA repair deficiency only rarely take a mutation in a Dna repair gene, but they instead tend to have epigenetic alterations such as promoter methylation that inhibit Dna repair gene expression.[nine] About 13% of colorectal cancers are deficient in Dna mismatch repair, commonly due to loss of MLH1 (9.viii%), or sometimes MSH2, MSH6 or PMS2 (all ≤1.5%).[10] For most MLH1-scarce sporadic colorectal cancers, the deficiency was due to MLH1 promoter methylation.[10] Other cancer types accept higher frequencies of MLH1 loss (meet tabular array below), which are once again largely a outcome of methylation of the promoter of the MLH1 gene. A different epigenetic machinery underlying MMR deficiencies might involve over-expression of a microRNA, for instance miR-155 levels inversely correlate with expression of MLH1 or MSH2 in colorectal cancer.[11]
| Cancer type | Frequency of deficiency in cancer | Frequency of deficiency in adjacent field defect |
|---|---|---|
| Stomach | 32%[12] [xiii] | 24%-28% |
| Stomach (foveolar type tumors) | 74%[xiv] | 71% |
| Breadbasket in high-incidence Kashmir Valley | 73%[15] | 20% |
| Esophageal | 73%[xvi] | 27% |
| Head and neck squamous prison cell carcinoma (HNSCC) | 31%-33%[17] [xviii] | xx%-25% |
| Non-small cell lung cancer (NSCLC) | 69%[19] | 72% |
| Colorectal | ten%[10] |
MMR failures in field defects [edit]
A field defect (field cancerization) is an area of epithelium that has been preconditioned by epigenetic or genetic changes, predisposing it towards development of cancer. Every bit pointed out past Rubin " ...there is bear witness that more than eighty% of the somatic mutations institute in mutator phenotype human being colorectal tumors occur before the onset of last clonal expansion."[20] [21] Similarly, Vogelstein et al.[22] signal out that more one-half of somatic mutations identified in tumors occurred in a pre-neoplastic phase (in a field defect), during growth of obviously normal cells.
MLH1 deficiencies were common in the field defects (histologically normal tissues) surrounding tumors; run across Table to a higher place. Epigenetically silenced or mutated MLH1 would probable not confer a selective reward upon a stem jail cell, however, it would cause increased mutation rates, and one or more than of the mutated genes may provide the cell with a selective advantage. The deficientMLH1 gene could then exist carried along as a selectively nearly-neutral passenger (hitch-hiker) gene when the mutated stem cell generates an expanded clone. The continued presence of a clone with an epigenetically repressed MLH1 would continue to generate further mutations, some of which could produce a tumor.
MMR components in humans [edit]
In humans, seven Dna mismatch repair (MMR) proteins (MLH1, MLH3, MSH2, MSH3, MSH6, PMS1 and PMS2) work coordinately in sequential steps to initiate repair of Dna mismatches.[23] In addition, there are Exo1-dependent and Exo1-contained MMR subpathways.[24]
Other gene products involved in mismatch repair (subsequent to initiation by MMR genes) in humans include Dna polymerase delta, PCNA, RPA, HMGB1, RFC and Dna ligase I, plus histone and chromatin modifying factors.[25] [26]
In sure circumstances, the MMR pathway may recruit an error-prone DNA polymerase eta (POLH). This happens in B-lymphocytes during somatic hypermutation, where POLH is used to introduce genetic variation into antibody genes.[27] However, this fault-prone MMR pathway may be triggered in other types of human cells upon exposure to genotoxins [28] and indeed it is broadly active in diverse homo cancers, causing mutations that bear a signature of POLH activity.[29]
MMR and mutation frequency [edit]
Recognizing and repairing mismatches and indels is of import for cells considering failure to do so results in microsatellite instability (MSI) and an elevated spontaneous mutation rate (mutator phenotype). In comparing to other cancer types, MMR-deficient (MSI) cancer has a very loftier frequency of mutations, close to melanoma and lung cancer,[xxx] cancer types caused by much exposure to UV radiation and mutagenic chemicals.
In addition to a very high mutation burden, MMR deficiencies consequence in an unusual distribution of somatic mutations across the human genome: this suggests that MMR preferentially protects the gene-rich, early on-replicating euchromatic regions.[31] In contrast, the gene-poor, belatedly-replicating heterochromatic genome regions exhibit loftier mutation rates in many human tumors.[32]
The histone modification H3K36me3, an epigenetic marker of active chromatin, has the ability to recruit the MSH2-MSH6 (hMutSα) complex.[33] Consistently, regions of the human being genome with high levels of H3K36me3 accumulate less mutations due to MMR action.[29]
Loss of multiple Dna repair pathways in tumors [edit]
Lack of MMR oftentimes occurs in coordination with loss of other DNA repair genes.[ix] For example, MMR genes MLH1 and MLH3 as well as 11 other DNA repair genes (such as MGMT and many NER pathway genes) were significantly downward-regulated in lower grade as well as in higher class astrocytomas, in contrast to normal encephalon tissue.[34] Moreover, MLH1 and MGMT expression was closely correlated in 135 specimens of gastric cancer and loss of MLH1 and MGMT appeared to be synchronously accelerated during tumor progression.[35]
Deficient expression of multiple DNA repair genes is often plant in cancers,[9] and may contribute to the thousands of mutations commonly found in cancers (see Mutation frequencies in cancers).
Crumbling [edit]
A popular idea, that has failed to proceeds significant experimental back up, is the idea that mutation, equally distinct from Dna damage, is the primary cause of crumbling. Mice lacking in the mutL homolog Pms2 have about a 100-fold elevated mutation frequency in all tissues, only practise non announced to age more apace.[36] These mice display by and large normal development and life, except for early onset carcinogenesis and male person infertility.
Meet also [edit]
- Base excision repair
- Nucleotide excision repair
References [edit]
- ^ Iyer RR, Pluciennik A, Burdett V, Modrich PL (February 2006). "Deoxyribonucleic acid mismatch repair: functions and mechanisms". Chemical Reviews. 106 (2): 302–23. doi:10.1021/cr0404794. PMID 16464007.
- ^ Larrea AA, Lujan SA, Kunkel TA (May 2022). "SnapShot: Dna mismatch repair". Jail cell. 141 (iv): 730–730.e1. doi:10.1016/j.prison cell.2010.05.002. PMID 20478261. S2CID 26969788.
- ^ Heller RC, Marians KJ (Dec 2006). "Replisome assembly and the directly restart of stalled replication forks". Nature Reviews. Molecular Cell Biology. 7 (12): 932–43. doi:ten.1038/nrm2058. PMID 17139333. S2CID 27666329.
- ^ Pluciennik A, Dzantiev Fifty, Iyer RR, Constantin Due north, Kadyrov FA, Modrich P (September 2022). "PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair". Proceedings of the National Academy of Sciences of the Us. 107 (37): 16066–71. doi:10.1073/pnas.1010662107. PMC2941292. PMID 20713735.
- ^ Kadyrov FA, Dzantiev L, Constantin Northward, Modrich P (July 2006). "Endonucleolytic function of MutLalpha in human mismatch repair". Cell. 126 (2): 297–308. doi:10.1016/j.cell.2006.05.039. PMID 16873062. S2CID 15643051.
- ^ Flores-Rozas H, Clark D, Kolodner RD (November 2000). "Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an agile mispair recognition circuitous". Nature Genetics. 26 (3): 375–8. doi:ten.1038/81708. PMID 11062484. S2CID 20861705.
- ^ Iyer RR, Pohlhaus TJ, Chen Southward, Hura GL, Dzantiev Fifty, Beese LS, Modrich P (May 2008). "The MutSalpha-proliferating cell nuclear antigen interaction in human Deoxyribonucleic acid mismatch repair". The Journal of Biological Chemistry. 283 (19): 13310–ix. doi:10.1074/jbc.M800606200. PMC2423938. PMID 18326858.
- ^ Online Mendelian Inheritance in Man (OMIM): 276300
- ^ a b c Bernstein C, Bernstein H (May 2022). "Epigenetic reduction of DNA repair in progression to gastrointestinal cancer". Globe Journal of Gastrointestinal Oncology. 7 (five): 30–46. doi:10.4251/wjgo.v7.i5.30. PMC4434036. PMID 25987950.
- ^ a b c Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, et al. (May 2005). "Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer". Gastroenterology. 128 (5): 1160–71. doi:10.1053/j.gastro.2005.01.056. PMID 15887099.
- ^ Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, et al. (April 2022). "Modulation of mismatch repair and genomic stability by miR-155". Proceedings of the National Academy of Sciences of the The states. 107 (xv): 6982–7. Bibcode:2010PNAS..107.6982V. doi:ten.1073/pnas.1002472107. PMC2872463. PMID 20351277.
- ^ Kupčinskaitė-Noreikienė R, Skiecevičienė J, Jonaitis L, Ugenskienė R, Kupčinskas J, Markelis R, et al. (2013). "CpG island methylation of the MLH1, MGMT, DAPK, and CASP8 genes in cancerous and adjacent noncancerous tum tissues". Medicina. 49 (8): 361–half-dozen. doi:10.3390/medicina49080056. PMID 24509146.
- ^ Waki T, Tamura Thou, Tsuchiya T, Sato Grand, Nishizuka S, Motoyama T (August 2002). "Promoter methylation status of E-cadherin, hMLH1, and p16 genes in nonneoplastic gastric epithelia". The American Journal of Pathology. 161 (two): 399–403. doi:10.1016/S0002-9440(10)64195-viii. PMC1850716. PMID 12163364.
- ^ Endoh Y, Tamura Grand, Ajioka Y, Watanabe H, Motoyama T (September 2000). "Frequent hypermethylation of the hMLH1 cistron promoter in differentiated-type tumors of the stomach with the gastric foveolar phenotype". The American Periodical of Pathology. 157 (3): 717–22. doi:x.1016/S0002-9440(ten)64584-1. PMC1949419. PMID 10980110.
- ^ Wani M, Afroze D, Makhdoomi M, Hamid I, Wani B, Bhat G, et al. (2012). "Promoter methylation status of DNA repair factor (hMLH1) in gastric carcinoma patients of the Kashmir valley" (PDF). Asian Pacific Journal of Cancer Prevention. xiii (8): 4177–81. doi:10.7314/apjcp.2012.13.8.4177. PMID 23098428.
- ^ Chang Z, Zhang W, Chang Z, Song M, Qin Y, Chang F, et al. (Jan 2022). "Expression characteristics of FHIT, p53, BRCA2 and MLH1 in families with a history of oesophageal cancer in a region with a high incidence of oesophageal cancer". Oncology Letters. 9 (1): 430–436. doi:x.3892/ol.2014.2682. PMC4246613. PMID 25436004.
- ^ Tawfik HM, El-Maqsoud NM, Hak BH, El-Sherbiny YM (2011). "Caput and neck squamous jail cell carcinoma: mismatch repair immunohistochemistry and promoter hypermethylation of hMLH1 cistron". American Journal of Otolaryngology. 32 (6): 528–36. doi:ten.1016/j.amjoto.2010.11.005. PMID 21353335.
- ^ Zuo C, Zhang H, Spencer HJ, Vural E, Suen JY, Schichman SA, et al. (Oct 2009). "Increased microsatellite instability and epigenetic inactivation of the hMLH1 factor in head and neck squamous cell carcinoma". Otolaryngology–Head and Neck Surgery. 141 (4): 484–90. doi:x.1016/j.otohns.2009.07.007. PMID 19786217. S2CID 8357370.
- ^ Safar AM, Spencer H, Su X, Coffey M, Cooney CA, Ratnasinghe LD, et al. (June 2005). "Methylation profiling of archived non-pocket-sized jail cell lung cancer: a promising prognostic system". Clinical Cancer Research. 11 (12): 4400–five. doi:10.1158/1078-0432.CCR-04-2378. PMID 15958624.
- ^ Rubin H (March 2022). "Fields and field cancerization: the preneoplastic origins of cancer: asymptomatic hyperplastic fields are precursors of neoplasia, and their progression to tumors can be tracked by saturation density in civilisation". BioEssays. 33 (iii): 224–31. doi:10.1002/bies.201000067. PMID 21254148. S2CID 44981539.
- ^ Tsao JL, Yatabe Y, Salovaara R, Järvinen HJ, Mecklin JP, Aaltonen LA, et al. (February 2000). "Genetic reconstruction of individual colorectal tumor histories". Proceedings of the National Academy of Sciences of the United States of America. 97 (three): 1236–41. Bibcode:2000PNAS...97.1236T. doi:10.1073/pnas.97.3.1236. PMC15581. PMID 10655514.
- ^ Vogelstein B, Papadopoulos North, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (March 2022). "Cancer genome landscapes". Science. 339 (6127): 1546–58. Bibcode:2013Sci...339.1546V. doi:10.1126/science.1235122. PMC3749880. PMID 23539594.
- ^ Pal T, Permuth-Wey J, Sellers TA (August 2008). "A review of the clinical relevance of mismatch-repair deficiency in ovarian cancer". Cancer. 113 (4): 733–42. doi:x.1002/cncr.23601. PMC2644411. PMID 18543306.
- ^ Goellner EM, Putnam CD, Kolodner RD (August 2022). "Exonuclease i-dependent and independent mismatch repair". DNA Repair. 32: 24–32. doi:10.1016/j.dnarep.2015.04.010. PMC4522362. PMID 25956862.
- ^ Li GM (January 2008). "Mechanisms and functions of DNA mismatch repair". Cell Inquiry. 18 (one): 85–98. doi:x.1038/cr.2007.115. PMID 18157157.
- ^ Li GM (July 2022). "New insights and challenges in mismatch repair: getting over the chromatin hurdle". Deoxyribonucleic acid Repair. xix: 48–54. doi:x.1016/j.dnarep.2014.03.027. PMC4127414. PMID 24767944.
- ^ Chahwan R, Edelmann W, Scharff MD, Roa S (August 2022). "AIDing antibiotic diversity past mistake-prone mismatch repair". Seminars in Immunology. 24 (4): 293–300. doi:10.1016/j.smim.2012.05.005. PMC3422444. PMID 22703640.
- ^ Hsieh P (September 2022). "DNA mismatch repair: Dr. Jekyll and Mr. Hyde?". Molecular Jail cell. 47 (5): 665–6. doi:10.1016/j.molcel.2012.08.020. PMC3457060. PMID 22980456.
- ^ a b Supek F, Lehner B (July 2022). "Clustered Mutation Signatures Reveal that Error-Prone Deoxyribonucleic acid Repair Targets Mutations to Agile Genes". Prison cell. 170 (3): 534–547.e23. doi:10.1016/j.cell.2017.07.003. hdl:10230/35343. PMID 28753428.
- ^ Tuna Chiliad, Amos CI (Nov 2022). "Genomic sequencing in cancer". Cancer Letters. 340 (2): 161–70. doi:10.1016/j.canlet.2012.11.004. PMC3622788. PMID 23178448.
- ^ Supek F, Lehner B (May 2022). "Differential Deoxyribonucleic acid mismatch repair underlies mutation rate variation across the human being genome". Nature. 521 (7550): 81–4. Bibcode:2015Natur.521...81S. doi:x.1038/nature14173. PMC4425546. PMID 25707793.
- ^ Schuster-Böckler B, Lehner B (August 2022). "Chromatin system is a major influence on regional mutation rates in human being cancer cells". Nature. 488 (7412): 504–7. Bibcode:2012Natur.488..504S. doi:ten.1038/nature11273. PMID 22820252. S2CID 205229634.
- ^ Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM (Apr 2022). "The histone mark H3K36me3 regulates human Deoxyribonucleic acid mismatch repair through its interaction with MutSα". Cell. 153 (iii): 590–600. doi:10.1016/j.jail cell.2013.03.025. PMC3641580. PMID 23622243.
- ^ Jiang Z, Hu J, Li X, Jiang Y, Zhou W, Lu D (December 2006). "Expression analyses of 27 Deoxyribonucleic acid repair genes in astrocytoma by TaqMan depression-density array". Neuroscience Letters. 409 (ii): 112–7. doi:x.1016/j.neulet.2006.09.038. PMID 17034947.
- ^ Kitajima Y, Miyazaki K, Matsukura South, Tanaka M, Sekiguchi M (2003). "Loss of expression of Deoxyribonucleic acid repair enzymes MGMT, hMLH1, and hMSH2 during tumor progression in gastric cancer". Gastric Cancer. 6 (2): 86–95. doi:x.1007/s10120-003-0213-z. PMID 12861399.
- ^ Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (Apr 1997). "Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2". Proceedings of the National Academy of Sciences of the Usa of America. 94 (7): 3122–seven. doi:10.1073/pnas.94.seven.3122. PMC20332. PMID 9096356.
Further reading [edit]
- Hsieh P, Yamane G (2008). "Dna mismatch repair: molecular mechanism, cancer, and ageing". Mechanisms of Ageing and Development. 129 (7–viii): 391–407. doi:x.1016/j.mad.2008.02.012. PMC2574955. PMID 18406444.
- Iyer RR, Pluciennik A, Burdett Five, Modrich PL (Feb 2006). "DNA mismatch repair: functions and mechanisms". Chemical Reviews. 106 (2): 302–23. doi:10.1021/cr0404794. PMID 16464007.
- Joseph N, Duppatla Five, Rao DN (2006). Prokaryotic Deoxyribonucleic acid mismatch repair. Progress in Nucleic Acid Inquiry and Molecular Biology. Vol. 81. pp. 1–49. doi:ten.1016/S0079-6603(06)81001-9. ISBN9780125400817. PMID 16891168.
- Yang W (August 2000). "Structure and office of mismatch repair proteins". Mutation Research. 460 (3–iv): 245–56. doi:10.1016/s0921-8777(00)00030-vi. PMID 10946232.
- Griffiths JF, Gilbert WM, Lewontin RC, Wessler SR, Suzuki DT, Miller JH (2004). An introduction to genetic analysis (eighth ed.). New York, NY: Freeman. ISBN978-0-7167-4939-four.
- Kunkel TA, Erie DA (2005). "Deoxyribonucleic acid mismatch repair". Annual Review of Biochemistry. 74: 681–710. doi:10.1146/annurev.biochem.74.082803.133243. PMID 15952900.
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger (2005). DNA repair and mutagenesis (second ed.). Washington, D.C.: ASM Press. ISBN978-i-55581-319-two.
External links [edit]
- DNA Repair
- Deoxyribonucleic acid+Mismatch+Repair at the US National Library of Medicine Medical Field of study Headings (MeSH)

Source: https://en.wikipedia.org/wiki/DNA_mismatch_repair
Posted by: stoltejoyagoint.blogspot.com
0 Response to "Would A Mismatch Repair Result In More Mutations If Using Newly Sunthesized Strand As A Template"
Post a Comment